|
A recent paper in Nature Communications (Abadi et al. 2019) investigated model selection in phylogeny reconstruction. Selecting an appropriate model of nucleotide substitution is considered best practice in phylogenetics, and indeed many studies have show that accurate modelling of substitution processes can substantially improve phylogenetic estimates. The authors quite surprisingly find that this practice may not be necessary after all. From multiple datasets of diverse simulated sequences, they find that the models chosen by commonly used criteria do not perform better than the most complex model. They conclude that cases model selection can be skipped altogether, and all phylogenetic inferences be performed with a complex model.
UPDATE (November 25, 2019): What is discussed here, together with some more in depth analyses, are now available as preprint here. There are other interesting aspects of this paper, but I here want to focus on the claim (and that data that's used to support it) that model selection does not improve phylogenetic inference. Below, I will argue that
7 Comments
Before writing about science again (new post is in the making), I have uploaded two scripts that I find useful for my work: gc_cov.pl This script is basically a very simplified version of the blobplots function from the BlobTools package. It creates GC-coverage plots directly from SPAdes assembly files, without the need for mapping the reads back to the assembly. Since I use mainly SPAdes anyway, this has been quite handy. The script will also annotate the plot when a taxonomy file is provided, which can be generated, e.g., from blast outputs. Below is an example for the plots that can be generated with the script. 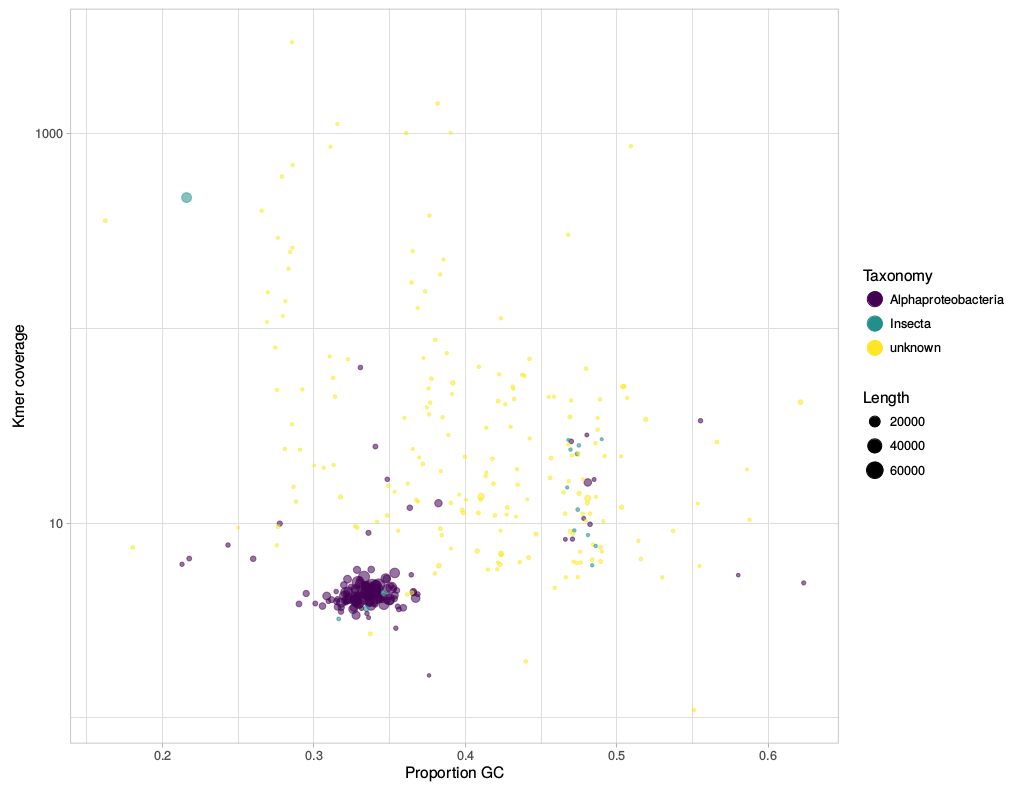 This post deals with a recent paper about Wolbachia in plant parasitic nematodes, and with Wolbachia phylogeny in general. Background Almost 30 genomes of the bacterial endosymbiont Wolbachia have been sequenced so far, and this trend is likely to continue. Wolbachia are found in a large proportion of arthropods (insects, arachnids, and allies) and in filarial nematodes. Very generally speaking, Wolbachia in arthropods are opportunistic, with varied fitness effects for their hosts, and may switch hosts horizontally. In contrast, Wolbachia in filarials are highly specialized and absolutely required for their hosts (the mechanisms underlying this co-dependence are not 100% clear yet). The differences in lifestyle of Wolbachia from arthropods and filarials is also reflected in their genomic architecture. For example, arthropod Wolbachia typically harbour many mobile genetic elements (e.g, insertion sequences, prophages & other phage-derived elements) that are almost always missing in the very streamlined and reduced filarial Wolbachia genomes. Now, for the first time, there is genomic data from more 'exotic' Wolbachia strains: Brown et al. have sequenced the genome of Wolbachia from a plant-parasitic nematode (wPpe from Pratylenchus penetrans), and, in a recent publication (Brown et al. 2016) compare it to the rest of the genomes of Wolbachia from arthropods and filarial nematodes. They also include in their analysis a strain from the banana aphid (wPni from Pentalonia nigronervosa) and a springtail (wFol from Folsomia candida). These strains were sequenced previously (De Clerck et al. 2015 & Gerth et al. 2014, respectively), but never investigated in a comparative framework before. All three strains are genetically very divergent from typical arthropod and filarial Wolbachia, so it was really cool to see this analysis published. Here, I want to briefly summarize the main findings of Brown et al.' s study and comment on what phylogenomic datasets and gene repertoires can tell us about evolutionary relationships within Wolbachia. In my first blog post, I will discuss a recent paper about Wolbachia classification. In a recent study, Wang et al. (2016) investigated Wolbachia sequences from cave spiders (Telema ssp.). They found that these belong to a genetic lineage distinct from all other described Wolbachia strains (in Wolbachia, those genetically distinct lineages are called “supergroups”). I re-analysed these data and found that in fact, Wolbachia strains from cave spiders cluster within supergroup A (Gerth 2016). [If you are unfamiliar with Wolbachia biology, or the supergroup classification system, the excellent review by Werren et al. (2008) is a good starting point.] After I uploaded my re-analysis to bioRxiv, Guan-Hong Wang has kindly send me the alignment files they used in their study. In this post, I want to try to use these data and illustrate why their and my analysis are discordant and also, why their conclusions are likely misled. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed