|
This post deals with a recent paper about Wolbachia in plant parasitic nematodes, and with Wolbachia phylogeny in general. Background Almost 30 genomes of the bacterial endosymbiont Wolbachia have been sequenced so far, and this trend is likely to continue. Wolbachia are found in a large proportion of arthropods (insects, arachnids, and allies) and in filarial nematodes. Very generally speaking, Wolbachia in arthropods are opportunistic, with varied fitness effects for their hosts, and may switch hosts horizontally. In contrast, Wolbachia in filarials are highly specialized and absolutely required for their hosts (the mechanisms underlying this co-dependence are not 100% clear yet). The differences in lifestyle of Wolbachia from arthropods and filarials is also reflected in their genomic architecture. For example, arthropod Wolbachia typically harbour many mobile genetic elements (e.g, insertion sequences, prophages & other phage-derived elements) that are almost always missing in the very streamlined and reduced filarial Wolbachia genomes. Now, for the first time, there is genomic data from more 'exotic' Wolbachia strains: Brown et al. have sequenced the genome of Wolbachia from a plant-parasitic nematode (wPpe from Pratylenchus penetrans), and, in a recent publication (Brown et al. 2016) compare it to the rest of the genomes of Wolbachia from arthropods and filarial nematodes. They also include in their analysis a strain from the banana aphid (wPni from Pentalonia nigronervosa) and a springtail (wFol from Folsomia candida). These strains were sequenced previously (De Clerck et al. 2015 & Gerth et al. 2014, respectively), but never investigated in a comparative framework before. All three strains are genetically very divergent from typical arthropod and filarial Wolbachia, so it was really cool to see this analysis published. Here, I want to briefly summarize the main findings of Brown et al.' s study and comment on what phylogenomic datasets and gene repertoires can tell us about evolutionary relationships within Wolbachia. What makes the wPpe genome special? I think the authors did a really good job in trying to answer that question. They looked at gene content, gene lengths, GC-content, coding density, mobile elements, conserved metabolic pathways, and many more things in the analysed genomes. They find that wPpe is in some regards very similar to the filarial nematode Wolbachia, but also shows similarities to arthropod Wolbachia. In summary however, these very detailed and thorough genomic analyses do not help to understand what might differentiate Wolbachia in plant-parasitic nematodes from the other Wolbachia strains functionally. There is no conspicuous difference in genomic architecture, or any metabolic functions present only in wPpe (or wPni & wFol). Further experiments will be necessary to determine its potential role. Unfortunately, this may not happen any time soon, as the authors state that culturing the nematode is very challenging. So from what we know now from the Brown et al. analyses, the wPpe genome is special because of its 1) phylogenetic placement and 2) gene content. I want to briefly comment on both of these points. 1) Wolbachia phylogeny Brown et al. used 79 conserved loci to analyse Wolbachia relationships and find support for a phylogeny that has been consistently recovered in recent analyses (e.g., Gerth et al. 2014; Nikoh et al. 2014; Comandatore et al. 2015; Ramírez-Puebla et al. 2015): supergroups A and B (the 'typical' arthropod Wolbachia) are reciprocally monophyletic and sistergroup to a clade ((C,F),D) – i.e., most of the 'typical' filarial Wolbachia. (A,B,C,D,F) is sister to supergroup E (wFol from the springtail). Now, for the novel part: the newly analysed strain wPni was recovered as sistergroup to all of the above strains (although it was sometimes also recovered as sister to wPpe) and wPpe as sistergroup to all sequenced Wolbachia strains (their Figure 2, see Figure 1 below). In addition to being in agreement with previously published analyses, the support for this topology is high and relatively consistent across the many analyses that were performed. 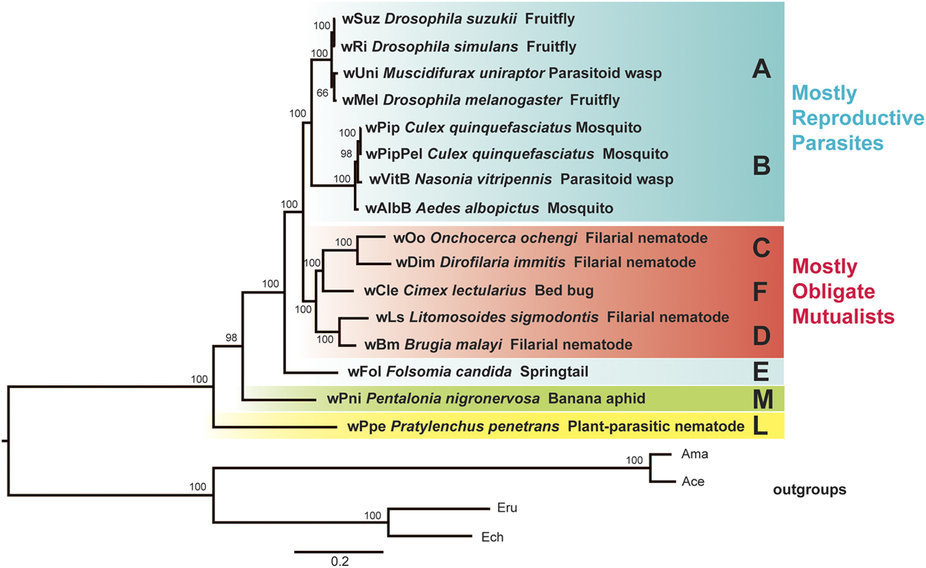 Figure 1: Wolbachia phylogeny based on 79 single copy orthologues (taken from Brown et al. 2016). Typical arthropod strains are highlighted in blue (supergroups A and B), typical filarial strains in red (supergroups C, D, F). The novel, 'exotic' strains are supergroups E, M, & L. It is therefore a bit peculiar that any published Wolbachia phylogeny paper, including this one, mentions long branch attraction (LBA) as a problem. Basically, the argument is that Wolbachia’s outgroups are separated from the ingroup by such a large phylogenetic distance that Wolbachia phylogenies tend to be distorted, with long branches being ‘drawn’ towards the root. This view is often found in the literature (one recent example: Lefoulon et al. 2016), and also prevalent on social media. I think this is largely due to the very influential paper by Seth Bordenstein and colleagues (Bordenstein et al. 2009), in which LBA is identified as a major problem in Wolbachia phylogeny. I like this paper, and absolutely agree with its conclusions, but much has changed since: we now have almost complete genomes from most Wolbachia supergroups and can pick those loci that are best suited for phylogenetics. Phylogenetic hypotheses can thus be tested with much more rigour and we can be more confident in our estimations. There is much less conflict in the data itself, which was not true for the Bordenstein et al. (2009) dataset. They came up with multiple conflicting phylogenies, strongly depending on the type of analysis that was performed (their Figs 1 & 3, see Fig. 2 below). If one reconstructs Wolbachia phylogeny with today's datasets (and follows best practices), one will always come up with the same (or at least a very similar) topology (Fig. 1). Just to be sure, I did another phylogenetic analyses with the novel genomes analysed by Brown et al., using a slightly different analytical aproach, and a larger dataset. As expected, I recovered the same topology (Fig. 3, see also methods summary below). 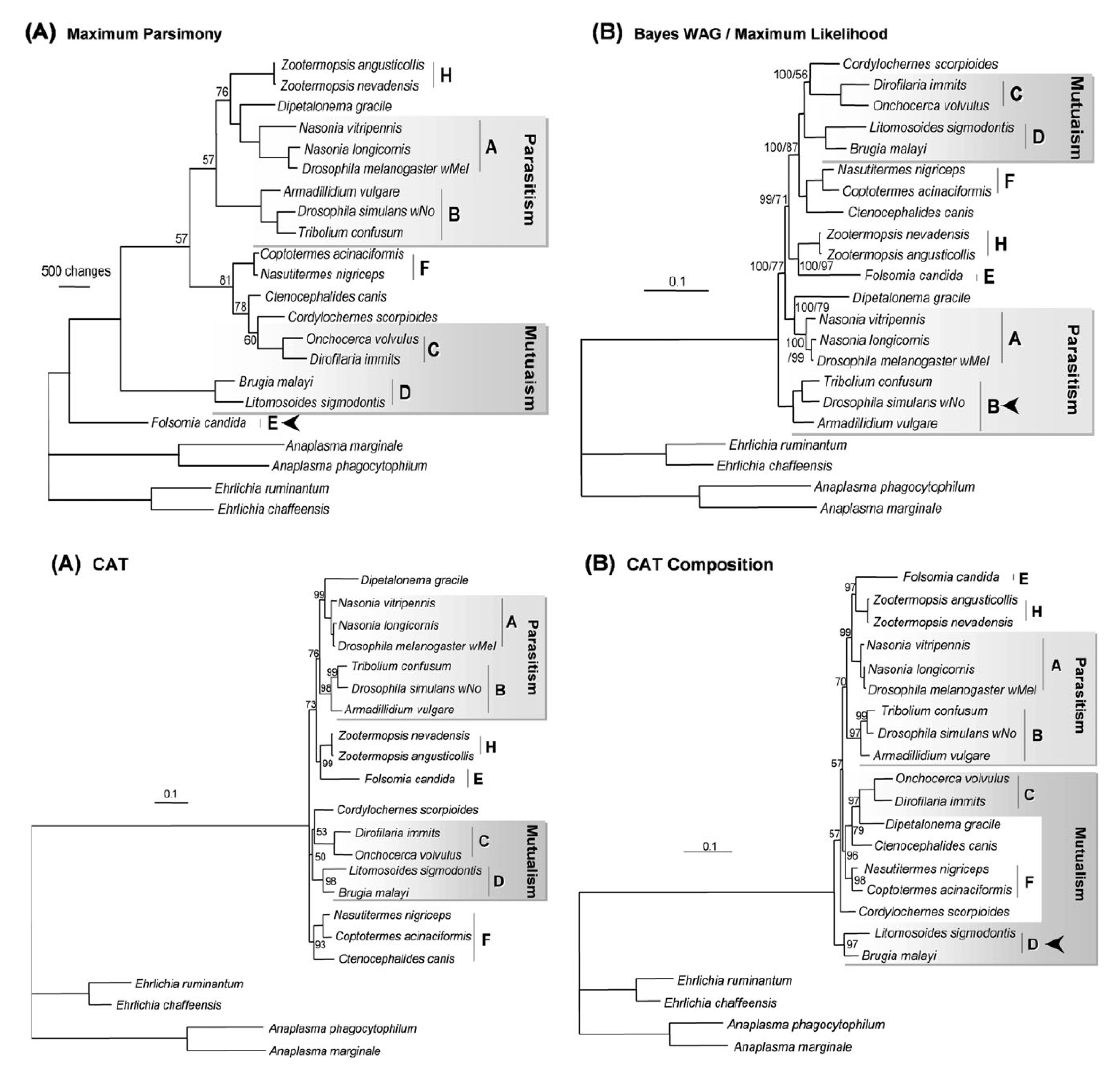 Figure 2: Wolbachia phylogeny based on 21 genes, taken from Bordenstein et al (2009, their Figures 1 & 3). Note how with this datset, support is generally low, and topologies change depending on the model / phylogenetic approach. This is very different for today's datasets derived from whole genomes. 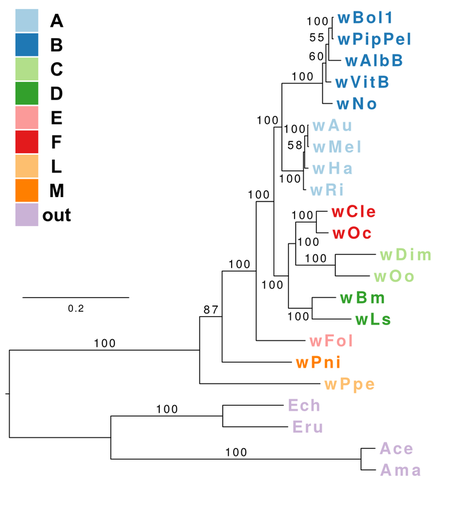 Figure 3: Maximum likelihood phylogeny of Wolbachia supergroups. Tree is based on partitioned amino acid supermatrix analysis with IQTREE (119 loci; 31,948 positions). PhyloBayes analysis (CAT-GTR, 2 chains, >20,000 generations) resulted in PP of 1 for all nodes, and the same tolopology exept for the placement of wPni (which was sistergroup to wPpe). Does that mean there is no LBA problem in Wolbachia phylogeny? No, not at all. LBA cannot objectively be proven or disproven for any real-world phylogenetic dataset, so this is of course also not possible for Wolbachia. I think however that the arguments for LBA as a big problem in this special case are not very strong. When we published our Wolbachia phylogeny (Gerth et al. 2014), and recovered wFol as sistergroup to the then sequenced strains, we faced the same criticism: our result was attributed to an LBA artifact, and more taxa would be needed to 'break' the long branch leading to the outgroups. Now, with the genomes of wPpe and wPni included in the analysis, the long branch is somewhat 'broken', and the placement of wFol has remained robust. I think that this argues against LBA. Another common argument is that 'all Wolbachia trees in which the longest branch is sistergroup to all other strains must be LBA artifacts'. But why can the 'true tree' not be just like this, with the longest branch at the root? It does not make sense to exclude a plausible tree only because a potential systematic error may have a similar appearence. So the question really is: Can we trust our data? When trying to control for phylogenetic biases and many types of systematic errors, a single Wolbachia phylogeny was recovered in multiple indenpendently performed analyses. When novel taxa are added (as in Brown et al.), the general topology remains robust. Therefore, I do not see strong reasons for doubt and I think that we now have a good idea about Wolbachia phylogeny. 2) Gene content analysis Another, somewhat independent line of evidence for this evolutionary scenario comes from gene content analyses. Fig. 4 shows an overview of shared orthogroups between Wolbachia strains. A great deal of loci are shared between all strains (shown in green), and also, there are many genes that are specific to a single supergroup (light blue). Then there are a number of gene clusters that have been lost in several lineages (light grey circles). Especially supergroup C and the strains wPni and wPpe are lacking many genes present in the other supergroups. 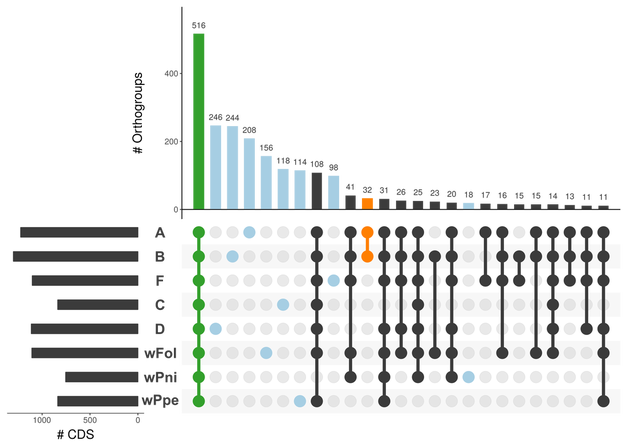 Figure 4: UpSet plot of orthogroups shared between Wolbachia strains. Each column stands for orthogroups shared between a number of strains. The size of the bars on top show how many orthogroups are shared (<10 not shown). Filled circles indicate which strains share these orthogroups, empty (light grey) circles indicate the absence of the orthogroups in the strain. Highlighted in green are orthogroups shared by all Wolbachia strains. Orthogroups found only in a single strain are highlighted in light blue. Finally, in orange, the orthogroups shared between supergroups A and B only are shown. Please note that for 'strains' A, B, C, D, F, the orthogroups of the pangenomes (i.e., orthogroups found in any strain of that Wolbachia supergroup) are shown. Barplot on the left shows the number of predicted CDS for each of the analysed strains. Interestingly, the only phylogenetic group that seems to be clearly supported by a number of newly acquired orthogroups is supergroups A+B (orange). For other phylogenetic lineages, evidence from gene content appears more ambiguous. Brown et al. argue that gene repertoire allies wPpe with supergroup C strains. They present a figure in support of this interpretation (Figure 5), which shows the proportion of genes that each analysed Wolbachia strain shares with wPpe. Of all analysed strains, wDim shares the largest proportion of genes with wPpe, so they conclude [...] gene repertoire analyses for single strains further support the association of the strains in plant-parasitic and filarial nematodes, particularly group C, with wPpe being the most similar to wDim. 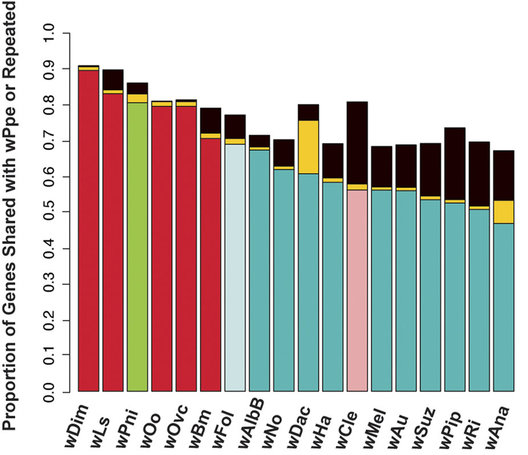 Figure 5 (taken from Brown et al. 2016): Proportion of genes from each analysed Wolbachia genome also found in wPpe. I think this representation is selective, and maybe a bit misleading. The problem is that supergroup C strains have very reduced genomes (as depicted in Figure 5), so their gene content is much closer to the Wolbachia core genome than that of other supergroups. In Fig. 6a, I have plotted the number (rather than the proportion) of orthogroups wDim shares with other Wolbachia strains and the outgroups. Evidently, wDim shares more genes with the outgroups than with wPpe. Following the above logic, one would have to argue that the outgroups are more closely related to wDim than most other Wolbachia strains, which is obviously incorrect. The graph below (Fig. 6b) shows the same thing for wMel. While the proportion of wMel genes found in wPpe is relatively low (Fig. 5), the number of wMel genes with orthologues in wPpe is even higher than the number for wDim genes found in wPpe! Again, this does not mean wMel is phylogenetically closer related to wPpe than wDim is. 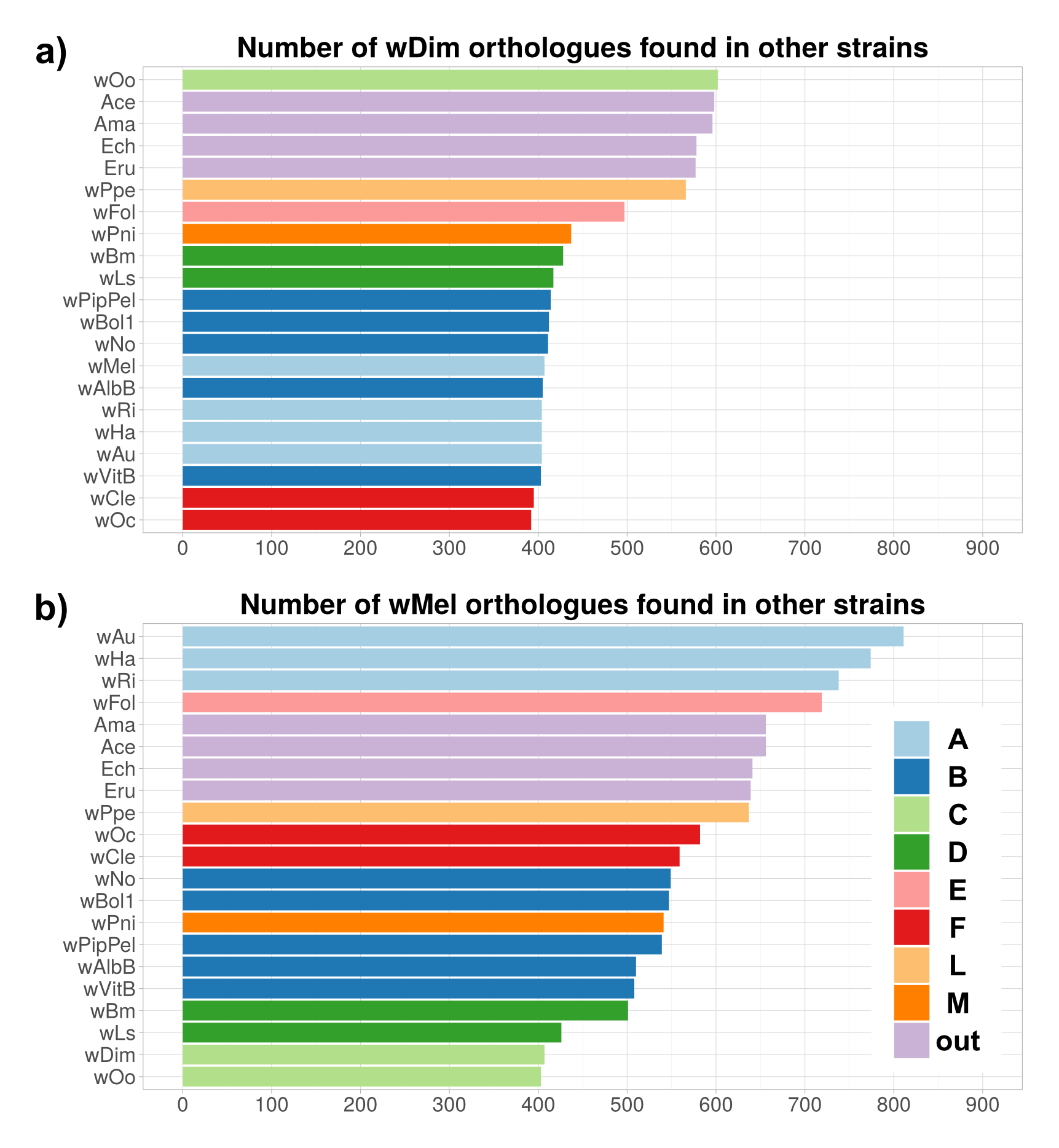 Figure 6: Number of wDim (a) and wMel (b) orthologues found in other strains. These examples illustrate that counting the number or proportion of shared genes is not a very good measure for phylogenetic relatedness. I think a better approach is to look at gene gain and loss in an evolutionary context. One way to do this is to create an absence/presence matrix for all orthogroups and to reconstruct a tree from this matrix in a maximum likelihood framework. When doing so, a tree is recovered that is more or less in agreement with the Wolbachia phylogeny recovered earlier (Fig. 7). The tree is not that well supported, and other topologies are likely not rejected by this orthogroup absence/presence dataset. However, the point I wanted to make here is that the gene repertoire of various Wolbachia strains is not in conflict (as Brown et al. seem to suggest), but rather supports evolutionary relationships estimated from whole genome nucleotide and amino acid datasets – especially regarding the placement of wPpe and wPni.  Figure 7: Maximum likelihood phylogeny of Wolbachia supergroups based on orthogroup absence/presence. Matrix was composed of 1523 positions (genes not assigned to orthogroups were excluded), and analysed with IQTREE (GTR2+FO+G4 model, 1000 bootstraps). Summary & conclusion With the novel Wolbachia genomes sequenced, recent previous estimations of Wolbachia evolution are largely corroborated. Support comes not only from analysis of single copy orthologues, but also from gene repertoire analysis. While this gives us a better understanding of Wolbachia evolution, the taxon sampling is probably still not dense enough to speculate about “the earliest Wolbachia hosts”. It'll be exciting to see genomic data of further, even more exotic strains! Methods summary All genomes were downloaded from public databases, wPni and wFol were assembled from raw reads with Megahit and SPAdes. Resulting contigs were annotated, and assemblies repeated with only the reads matching to Wolbachia contigs. This filtering was repeated until only contigs matching Wolbachia were found. All genomes were annotated with Prokka. Orthogroups were determined with Orthofinder. Amino acid dataset for phylogenetic analyses was assembled from single copy orthologs present in all analysed genomes. Recombining loci (identified with via pairwise homoplasy index and window sizes of 10, 20, 30, 40, and 50) and loci that were biased in amino acid composition were removed. Phylogenetic analyses were performed with IQTREE (partitioned supermatrix with best model and partitioning scheme determined beforehand) and PhyloBayes MPI (concatenated supermatrix, CAT-GTR model). Graphs were done in FigTree and R using ggplot2 and UpSetR; editing was done in with Inkscape. Data files wFol assembly wPni assembly protein supermatrix best partitioning scheme gene presence/absence matrix all predicted proteins of all genomes Orthofinder results References Bordenstein SR, Paraskevopoulos C, Dunning Hotopp JC, Sapountzis P, Lo N, Bandi C, Tettelin H, Werren JH, Bourtzis K (2009) Parasitism and mutualism in Wolbachia: what the phylogenomic trees can and cannot say. Molecular Biology and Evolution 26, 231–241. Brown AMV, Wasala SK, Howe DK, Peetz AB, Zasada IA, Denver DR (2016) Genomic evidence for plant-parasitic nematodes as the earliest Wolbachia hosts. Scientific Reports 6, 34955. Comandatore F, Cordaux R, Bandi C, Blaxter M, Darby A, Makepeace BL, Montagna M, Sassera D (2015) Supergroup C Wolbachia, mutualist symbionts of filarial nematodes, have a distinct genome structure. Open Biology 5, 150099. De Clerck C, Fujiwara A, Joncour P, Léonard S, Félix M-L, Francis F, Jijakli MH, Tsuchida T, Massart S (2015) A metagenomic approach from aphid’s hemolymph sheds light on the potential roles of co-existing endosymbionts. Microbiome 3, 63. Gerth M, Gansauge M-T, Weigert A, Bleidorn C (2014) Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nature Communications 5, 5117. Lefoulon E, Bain O, Makepeace BL, d’Haese C, Uni S, Martin C, Gavotte L (2016) Breakdown of coevolution between symbiotic bacteria Wolbachia and their filarial hosts. PeerJ 4, e1840. Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T (2014) Evolutionary origin of insect-Wolbachia nutritional mutualism. Proceedings of the National Academy of Sciences of the United States of America 111, 10257–10262. Ramírez-Puebla ST, Servín-Garcidueñas LE, Ormeño-Orrillo E, Vera-Ponce de León A, Rosenblueth M, Delaye L, Martínez J, Martínez-Romero E (2015) Species in Wolbachia? Proposal for the designation of “Candidatus Wolbachia bourtzisii”, “Candidatus Wolbachia onchocercicola”, “Candidatus Wolbachia blaxteri”, “Candidatus Wolbachia brugii”, “Candidatus Wolbachia taylori”, “Candidatus Wolbachia collembolicola” and “Candidatus Wolbachia multihospitum” for the different species within Wolbachia supergroups. Systematic and Applied Microbiology 38, 390–399.
2 Comments
4/12/2016 06:04:50
Thanks for the fun post and new analyses, Micheal! As we just discussed on Twitter, I think the presence/absence analysis is interesting though weakly supported as you note. LBA remains a strong explanation for the core gene phylogeny though IMO. 1. First the root is the longest branch in the Wolbachia ingroup. I wish it wasn't so we wouldn't have to worry about LBA :) 2. Second all three of the longest branches, including wFol, cluster together at the root. That's also a classic LBA effect. 3. We don't need more long branches to "break" up the root IMO but rather more closely related sister taxa to the long branches to potentially get them out of the long branch cluster at the root. As wFol shifts placement in the trees above, I suspect this may be the first to move from near the root to elsewhere once closer relatives to wFol are found. So in sum, while I want to find the real root, I think we're still far off from having confidence now. Heartily agree that more sampling will bring us closer to the answer! More sampling within long branches and in exotic species is needed - not a small task! We have much work to do.
Michael Gerth
19/12/2016 14:43:26
Thanks for the comment Seth! I should have mentioned that the phylogenetic estimates from gene content analyses are probably more biased than the ones from matrices of single copy genes. Gene loss is rampant in endosymbiotic bacteria, and it is very likely that many gene losses happen convergently (independently) in multiple lineages. Leave a Reply. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed