|
A recent paper in Nature Communications (Abadi et al. 2019) investigated model selection in phylogeny reconstruction. Selecting an appropriate model of nucleotide substitution is considered best practice in phylogenetics, and indeed many studies have show that accurate modelling of substitution processes can substantially improve phylogenetic estimates. The authors quite surprisingly find that this practice may not be necessary after all. From multiple datasets of diverse simulated sequences, they find that the models chosen by commonly used criteria do not perform better than the most complex model. They conclude that cases model selection can be skipped altogether, and all phylogenetic inferences be performed with a complex model.
UPDATE (November 25, 2019): What is discussed here, together with some more in depth analyses, are now available as preprint here. There are other interesting aspects of this paper, but I here want to focus on the claim (and that data that's used to support it) that model selection does not improve phylogenetic inference. Below, I will argue that
1) Short (& incomplete) summary of the data presented in the paper (skip this if you know the paper already)
The authors have compiled 7,200 nucleotide alignments from three different databases. These alignments cover a large range of sequence length and numbers, but are biased towards short sequences with few taxa (which makes sense for the scope of this paper, see figure below). From this dataset, 4 other datasets of equal size were simulated, which won't be discussed in detail in this post. The focus were 24 nucleotide substitution models (JC, F81, K2P, HKY, SYM, GTR, and each of those combined with estimation for the gamma parameter for rate heterogeneity and/or invariable sites), and six different approaches of determining the best fitting model (which also are not important in the context of this post). 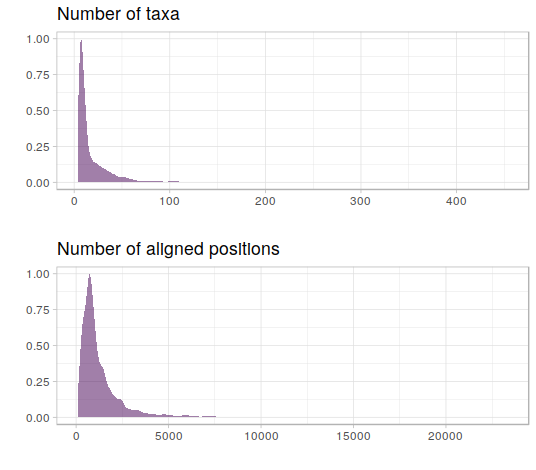
Features of alignments used in Abadi et al. (2019).
While the original aim of the study was to determine the best approach to chose models for phylogenetic reconstruction, it appeared that not only all of these approaches gave similar results in terms of phylogenetic accuracy, but also that choosing GTR+G+I performs comparable to phylogenetic reconstruction with the best fitting model.
But what data are used to support this claim? First, the authors reconstruct trees with the various "best fitting" models, and with GTR+G+I (and also JC) for each of the 7,200 alignments for all simulated datasets. They then count how often the chosen model returns the "true tree" (tree used for simulating the datasets). Here, all approaches result in ~50% recovery rate of the true tree (their Table 2 pasted below). 
Table 2 from Abadi et al. (2019).
Next, they measured Robinson-Foulds (RF) distances between the "true tree" and each of the trees recovered by applying the different best model determination approaches. They rank the RF distances from 1 (close to true tree) to 9 (distant from true tree), and report averages in their Table 3 (pasted below). Again, they find that all strategies perform very similar.
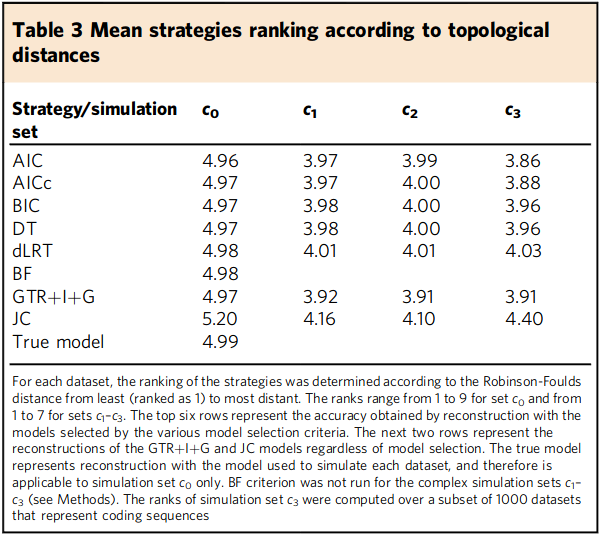
Table 3 from Abadi et al. (2019).
They also show the actual RF distances in Figure 4, again the impression is that they are all quite similar.
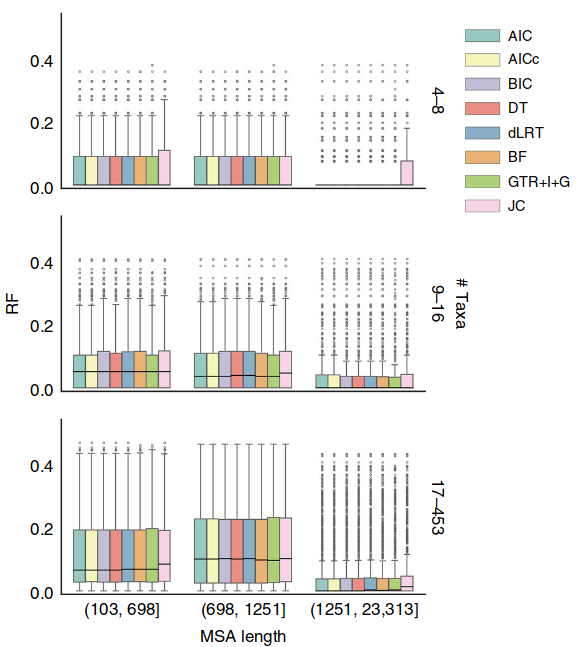
Figure 4 from Abadi et al. (2019).
2) What does and what doesn't this show?
Quite convincingly, the data demonstrate that on average, using the best model as determined by any criterion and using GTR+G+I without prior model choice results in similar trees. The main issue I see here is that the results are averaged over each of the datasets, and this may not be the best approach of testing the hypothesis 'Model choice does [not] matter for topology reconstruction'. The problem is that in practise, when reconstructing phylogenies, one is probably not so much interested in averages, but in the particular features of the alignment in question. For example, most people would likely agree that typically, GTR+G+I is the best choice for most datasets (from the models analysed in the paper). Model selection is still routinely performed because people also likely agree that not all datasets are typical/average. What I would need to see to abandon model choice is that GTR+G+I consistently performs equally well or better than any other model for any other dataset (or at least most other models in most datasets). The paper does not address this, and this is why I think the conclusions are too bold. For each of the different criteria investigated, the authors lump all best models together, without reporting the proportions of the actual models. Its impossible to see from this data if any of the approaches (AIC, BIC, GTR+G+I, JC, etc) consistently performs better or worse with any of the alignments. Thought experiment: lets assume that the JC model consistently gives more accurate estimates for alignments in which the JC model was determined as best fitting, and consistently worse estimates for alignments in which GTR+G+I was determined as best fitting. In this case, when averaging across all models this pattern could result in the same average values for tree distances as in the case when JC and GTR+G+I always give the same tree for any dataset. The conclusions would be quite different though; in the former case model choice would matter, in the latter case it wouldn't. We therefore cannot conclude from the data presented in the paper if model choice really is not necessary. 3) Re-analysis of the empirical dataset There are several aspects of data presentation in the paper that I think could be improved. As mentioned above, I think it would have been beneficial to look at the different models that were selected as best models instead of combining them all in the different criteria used for model choice. Further, in various places in the paper, the authors use the fraction of correctly recovered trees as measurement for accuracy. I think using actual RF distances throughout would have been beneficial. In some parts of the analysis, a tree that differs from the "best" tree in a single node is counted as equally wrong as a tree that differs in every single node. I feel this should be more nuanced. When actual RF distances are actually used (in Figure 4) they are heavily biased by 0 RF distances, so its very hard to determine any differences. Likewise, ranking RF distances instead of reporting actual values (as in Table 3) can obscure actual differences in RF distances. To investigate some of these issues, I downloaded the 7,200 alignments of the empirical dataset the authors have compiled. I then used ModelFinder implemented in IQ-TREE to determine the best models for each of the 7,200 alignments. I reconstructed the tree using the best models and also with GTR+G+I, and computed normalized RF distances with RAxML to compare between the resulting trees. First, how often was each of the models determined as best fitting? 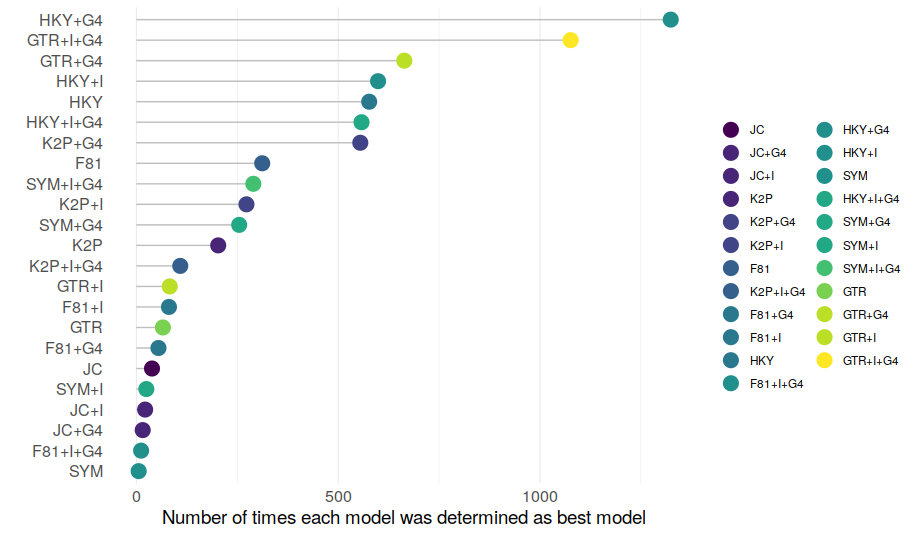
Number of alignments (out of 7,200) for which each model was chosen as best fitting by ModelFinder implemented in IQ-TREE (default options). Colour scale represents model complexitiy from JC (least complex) to GTR+I+G4 (most complex).
We can see that models were selected unequally often, which is not that surprising. In this dataset of 7,200 empirical alignments, around half of all models were recovered as best fitting at a non-negligible frequency, which shows that this is not a bad dataset to test if model selection matters.
Now, how often does GTR+G+I give identical trees to analyses using the best model? 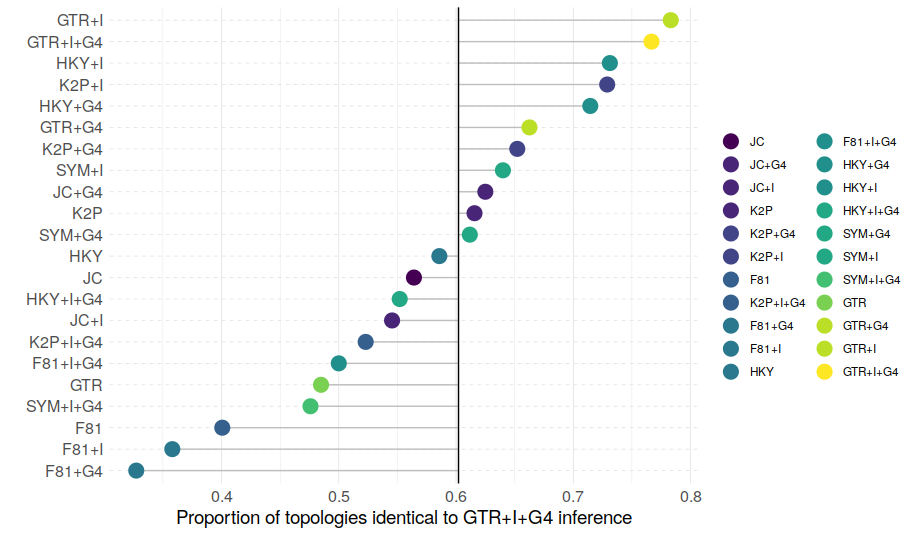
Proportion of trees from ML reconstruction with best fitting models that are identical to tree obtained with GTR+G+I model. Only models that were determined to be best fitting at least 10 times are shown. The vertical black line indicates the averaged value across all models.
The graph shows the proportion of trees that were found to be identical when using GTR+G+I and the best model. The central vertical line represents the mean across all alignments (this corresponds to the value that was reported in Abadi et al.'s Table 2 – see above).
GTR+G+I performs better than average for some alignments and considerably worse than average for others. There seems to be a trend towards trees calculated with more complex models being better recovered by GTR+G+I, but this trend is not very consistent. The F81 model trees are especially badly recovered with GTR+G+I. In summary, the empirical dataset compiled by the authors quite convincingly demonstrates that there is a large impact of model selection on phylogenetic accuracy. In other words, you get different trees when using GTR+G+I instead of the best model, and the difference depends on which model best describes the data. [Note that unlike in the analyses presented in the paper, the analyses here are not about the "true tree", this is unknown. What is shown is merely the difference between the trees under GTR+G+I and the best models.] Finally, for the trees that are not identical, how different are the GTR+G+I trees from the trees reconstructed with best models? 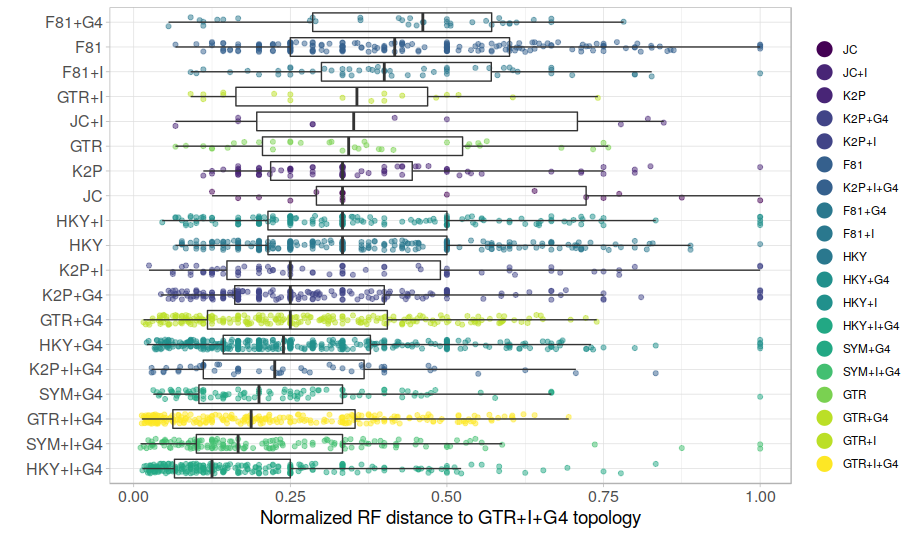
RF distances between ML trees recovered with GTR+G+I and with using the best models. Graph excludes 0 distances. Only models that were determined to be best fitting at least 10 times are shown.
This graph looks a bit messy but still shows that model choice does impact tree reconstruction. GTR+G+I produces more similar trees for some alignments than for others, and the degree of similarity depends on the best model of nucleotide substitution.
You may have noticed that even if the best model chosen was GTR+G+I, The RF distances can be relatively large. In other words, when using the same best model (GTR+G+I) twice, we can get quite different trees. This is not that surprising as such, but I would assume that if you do use the same best model for two independent ML analyses of the same alignment, and get very different trees, then there is probably a serious lack of phylogenetic signal in the alignment. I think this may be a reason why the above graph looks so messy. Lets look at the data again, but only include alignments for which two independent runs of the best model give tree estimates that are at most 10% different. In other words, lets only include alignments with more consistent phylogenetic signal. 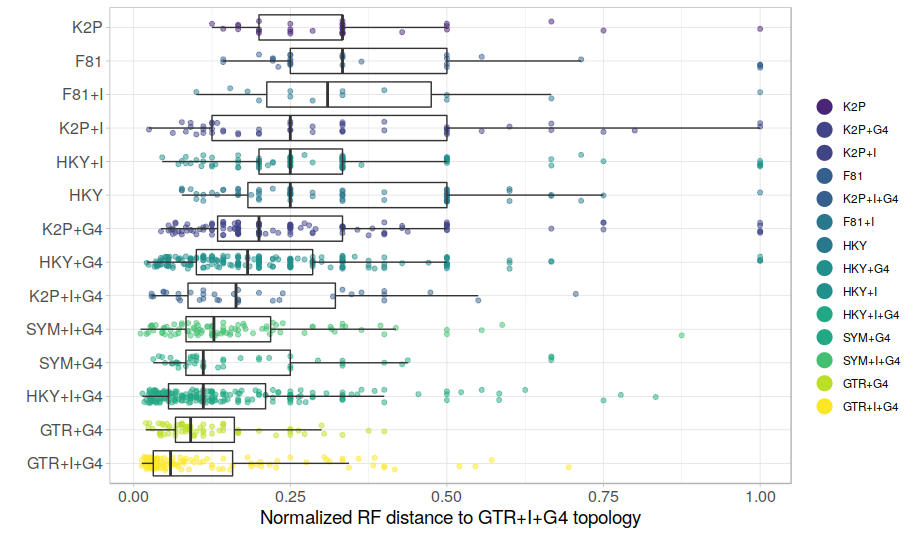
RF distances between reduced set of alignments with consistent phylogenetic signal (4673 or 65% out of 7,200 alignments).
This graph looks a bit more tidy, and the differences between models now appear even clearer. I think this is probably a better representation of the data, but it doesn't really change the conclusions.
To sum up the re-analysis of this dataset: model choice has a considerable impact on ML tree reconstruction, both in terms of proportion of equally recovered topologies and in terms of distances between unequally recovered topologies. Neither of these trends are obvious from the results presented in Abadi et al. (2019), and this is due to the unfortunate choice of showing only averaged results instead of focussing on differences between best models. One of the main conclusions of the paper is that model selection is not necessary. I believe that my brief re-analysis has shown that this conclusion is not strongly supported by the data brought forward (at least for the 'empirical' dataset). In reality, people might feel model selection is tedious and results in GTR+G+I in most cases anyway, and this is a fair point. The question is, what about the cases in which GTR+G+I is not the best fitting model, knowing that using GTR+G+I can lead to substantially different trees than the best fitting model? Would one be ready to trade accuracy and best practices for convenience? Consider testing for normality before choosing a suitable statistical approach as analogy: You may already know that the data are normally distributed by looking at them, or you have done many similar measurements in the past and the data have always been normally distributed. You would probably still test for normality, knowing that choosing an inappropriate downstream statistical analysis may result in erroneous results. 4) What is the benefit of skipping model selection anyway? Even if many models of nucleotide substitution perform similar in an ML framework, it is unclear what the benefit of skipping model selection would be. There is some very user-friendly software to do model selection that come with very good documentation (e.g., jModeltest or ModelFinder), so convenience is not really a good argument. The only argument brought forward by Abadi et al. (in addition to the claim that there is no benefit for phylogenetic accuracy) is that it would save CPU time to always use GTR+G+I by default. But is that really the case? After all, fitting more parameters for a complex model also comes at a computational cost. In the re-analysis of the 7,200 alignments, using GTR+G+I took on average 1.42 times longer than the time needed for looking for the best model and tree search under the best model, which means that its computationally more expensive to use GTR+G+I by default. This number comes with 2 caveats however: 1) Only 24 models were tested, and testing for the whole suite of available models likely takes a little longer. 2) All model searches were done under the default option in ModelFinder/IQ-TREE, which means that there was not a full tree search for each model, but rather all models were optimized with a parsimony tree. I was interested in what effect this quicker model search had on the accuracy, and therefore also performed full tree searches for all the alignments under all 24 models. The default option results in the exact same model for 82% (5,890) of alignments and in the same substitution matrix for 86% (6,225) of alignments. First, I checked if that changed any of the findings discussed above, and it did not. I think its likely that the "wrong" models chosen with the quick method still perform reasonably/similarly to the correct one, but I did not test this. For people who are concerned about the computational cost of model selection, the default model search in ModelFinder or the 'bionj' option in jModelTest might make good alternatives to full tree searches.
Summary
7 Comments
2/4/2019 09:39:40
Very nice post. It makes lot of sense. See also https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3175760/
lixingguang
25/12/2019 02:41:42
I agree with you, and if a dataset contains very little phylogenetic information, difference models give very different results.
Michael Gerth
2/4/2019 10:53:37
Thank you! And sorry for not giving due credit to this paper of yours (or any other studies really) in the post. My knowledge of the relevant literature is far from comprehensive, but my impression has always been that not all alignments behave equally and that accurate modelling of substitution processes does impact phylogenetic reconstruction. 2/4/2019 11:06:50
No worries, I just meant that other studies have looked at these questions before. Indeed, I agree with you. In many cases, particularly at high levels of divergence, GTR+I+G will be or will do as well as the best-fit model, but you never know about your particular dataset. It is safer and easy enough to do model selection. BTW, you should send your reply to NatComm!!
lixingguang
25/12/2019 02:32:06
I totally agree with you, and I really do not know why NatComm accept this kind of paper, just for different opinion from others?
Michael Gerth
2/4/2019 12:24:25
Re. It is safer and easy enough to do model selection – Yes, agreed 100%!
lixingguang
25/12/2019 02:47:34
I have made many comments about other papers and submitted to their published journals, includes Nature, they all rejected me without really response to me. So this is a very tough way for us, journal is journal, they are not science, this is what I get. Leave a Reply. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed