|
This blog post deals with the various ways of downloading large amounts of sequencing data (e.g., from NCBI’s SRA database). When I needed to bulk download short read for a recent project, it took me some time to figure out how to achieve this efficiently, and I am sharing my experience here in the hope it might be useful.
The problem: you want to download lots of sequencing data (typically in form of Illumina generated reads), e.g., to reproduce a published experiment. The amount of data makes it impossible to click+download through a browser interface. There are two potential solutions: 1) download via NCBI’s SRA toolkit, and 2) access ftp servers directly.
1) SRA Toolkit NCBI stores all submitted sequencing reads in the sra format. This format is used to archive and compress various sequence file formats. Here, I will only consider sra files that contain compressed fastq read files as created with Illumina base calling software. The biggest problem with the sra format is the lack of universality. The majority of software commonly used in downstream analyses of these reads cannot handle this format – in fact, none of the software that I use regularly recognizes sra files. So whenever you access the SRA database, you will have to download sra files, and then convert them into fastq files (often, one would also compress the fastq files with gzip or bzip2). A typical workflow would involve looking for the experiments of interest on the SRA webpage, and export the accession list (select runs by clicking on the boxes, then navigate to 'Send to' -> 'File' -> 'Accession List'). It also makes sense to download all metadata associated with the selected runs ('Send to' -> 'Run selector', and in the new window 'Download' -> 'RunInfo Table'). With the accession list (here called 'SraAccList.txt'), one can download the SRA files with the SRA Toolkit, a software developed by NCBI to access the SRA database and to manipulate sra files. Download the software here, extract the archive, and find all binaries in the 'bin' folder. To download the sra files associated with the accession numbers in the list, run
(Prefetch used to be able to read lists of sra accession numbers directly, but the new version doesn't seem to able to do that anymore, so I am here using xargs.) Unfortunately, prefetch will dump all files into where you very like don’t want them – your home folder. To change this behaviour, run
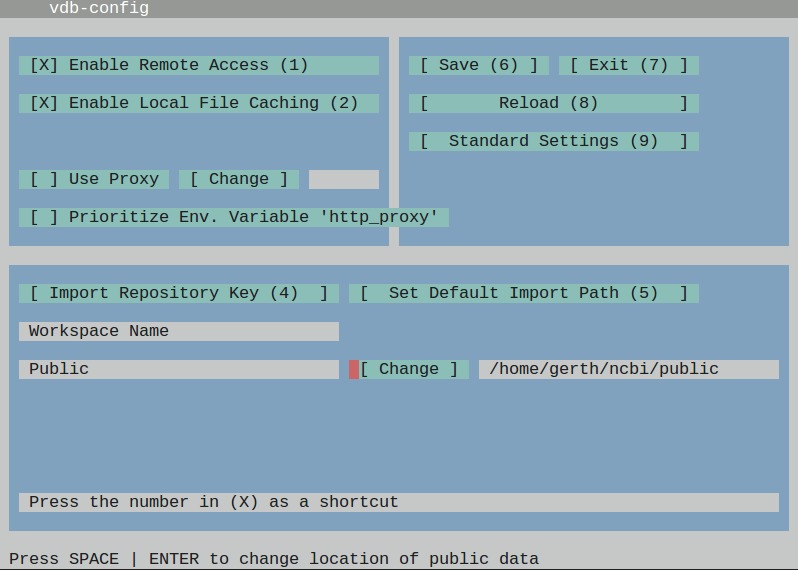
and use the graphical interface to alter the path under which the SRA files are stored by default (option number 5).
Next, the sra files can be converted to gzipped fastq files with
Alternatively, one can skip the prefetch command (i.e., combine the previous two commands into one) by calling
directly. Note that this will still download sra files and store them on your computer, so it won't actually be faster or more resource friendly. It therefore makes sense to regularly empty the sra directory after the conversion to fastq files is completed. Fastq-dump has many other options, not all of which are very intuitive. Please see this excellent blog post by Rob Edwards dealing with the most important ones. Summary The SRA toolkit is NCBI’s recommended and only supported way of accessing the SRA database. The main problem in my view is that the conversion from sra into fastq into gzipped fastq takes up a lot of time and computational resources. Depending on the hardware you are using, this can become problematic. Furthermore, I found the download speeds disappointing, and connections to the server are often interrupted. Another drawback is the documentation of the software, which seems unnecessarily complex. I only use the SRA Toolkit if I can't access the files I need via the method described below. 2) Ftp download As far as I know, there is no way of directly accessing fastq files from NCBI. However, there is a ftp server which can be accessed using wget or a browser. Having said that, this still leaves the problem of converting sra files into fastq. Luckily, the European Nucleotide Archive (ENA) stores these file types, and they can be accessed in a very convenient way. The ENA has all the files that can be found in NCBI’s SRA, but there is a time lag in uploading – you may not find the newest NCBI submissions on ENA. The ENA ftp server is organised in a directory structure that derives from the accession number itself. The structure is explained very well on the ENA homepage, and summarised in the scheme below: 
Once you know the ftp directory, the files can be directly downloaded using wget, e.g.:
Depending on the available bandwidth, this can be quite fast and is in my experience already faster than using the SRA toolkit, especially when considering that a conversion of sra files is not necessary. To achieve even greater download speeds however, I would advise on using the aspera download client. This is commercial, closed source software, which is not ideal – but its free to use and the download speeds are amazing. To use it, either download and install the aspera connect command line utility or the aspera connect browser plugin, which also will install the command line version alongside. The default installation directory is $HOME/.aspera/. To use aspera with ENA, three files are needed: the program itself ('ascp', installed to 'connect/bin/'), a license file ('aspera-license', in 'connect/etc/'), and an ssh key ('asperaweb_id_dsa.openssh', also in 'connect/etc'). It is probably most convenient to add the whole directory to your PATH variable. Accessing fastq files is very similar to what I described above, only the address is a bit different. Simply replace 'ftp://ftp.sra.ebi.ac.uk/' in the links above with '[email protected]:', the rest is identical. To download using aspera:
Here, -i points to the path of the openssh key, which always needs to be explicitly provided. Please read the documentation for an explanation of all options. Using ascp, typical download speeds I have observed are 100–500Mbps which is impressive. I also encountered connection problems sometimes, but generally more rarely than with the SRA toolkit. I should mention that aspera also works with prefetch from NCBI's SRA toolkit, which increases download speeds considerably compared to the 'regular' prefetch. Overall however, prefetch with aspera is still much slower than direct dowload from ftp using ascp. Naturally, when downloading large amounts of sequencing data, one doesn’t want to type endless wget or ascp commands. Given the rules outlined above, one can infer the ftp location of all fastq files on the ENA ftp server from their accession numbers, which should be straightforward to automate. In case you don’t want to do this yourself, please feel free to try out the script I created for this purpose. It reads in a list of accession numbers and downloads the corresponding libraries from ENA using wget or ascp. Download the script and run
Summary This script, used with aspera, is now my preferred way of downloading fastq files from ENA. The downloads are very fast, both on the cluster I regularly use, and on my personal desktop computer. The only downside is that not all experiments from NCBI can be found on ENA – the newest submissions are often missing. Furthermore, the ENA servers are situated in Europe. I have no idea how this might influence download speeds in other parts of the world (it would be great to get feedback on this point). If you decide to give the script a try, please let me know how it works for you!
7 Comments
Juan Pascual Anaya
8/8/2018 01:37:45
This is a great post, very useful information. Downloading bulk SRA data is very painful, I wish NCBI provided some better tools, but both fastq-dump and the newer fasterq-dump are really bad (slow as hell). I'm using your Perl script with ascp and it's quite sweet. Thanks!
Michael Gerth
8/8/2018 13:34:51
Dear Juan, thanks for the kind words! I am happy if this is useful for you. I agree, using NCBI's sra-tools can be very frustrating. Luckily, ENA offers a less complicated way of accessing SRA data.
Zegang Wei
21/6/2019 17:45:19
Hi, I tried to download fastq file from DDBJ, but the error always coours:
Michael Gerth
2/7/2019 12:45:41
Hi Zegang, sorry, but this seems to be a problem specific to the DDBJ. I suggest you contact them to resolve this issue. Alternatively, download the libraries from ENA – they should be available there as well.
Karisma
26/11/2019 13:19:30
Can't thank you enough for this! One of the most helpful posts I've seen about accessing SRA/ENA.
Michael Gerth
26/11/2019 20:07:53
Thanks Karisma, happy you find this helpful!
Kristina
30/5/2024 14:19:01
Dear Michael Leave a Reply. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed