|
This blog post deals with the various ways of downloading large amounts of sequencing data (e.g., from NCBI’s SRA database). When I needed to bulk download short read for a recent project, it took me some time to figure out how to achieve this efficiently, and I am sharing my experience here in the hope it might be useful.
The problem: you want to download lots of sequencing data (typically in form of Illumina generated reads), e.g., to reproduce a published experiment. The amount of data makes it impossible to click+download through a browser interface. There are two potential solutions: 1) download via NCBI’s SRA toolkit, and 2) access ftp servers directly.
6 Comments
Before writing about science again (new post is in the making), I have uploaded two scripts that I find useful for my work: gc_cov.pl This script is basically a very simplified version of the blobplots function from the BlobTools package. It creates GC-coverage plots directly from SPAdes assembly files, without the need for mapping the reads back to the assembly. Since I use mainly SPAdes anyway, this has been quite handy. The script will also annotate the plot when a taxonomy file is provided, which can be generated, e.g., from blast outputs. Below is an example for the plots that can be generated with the script. 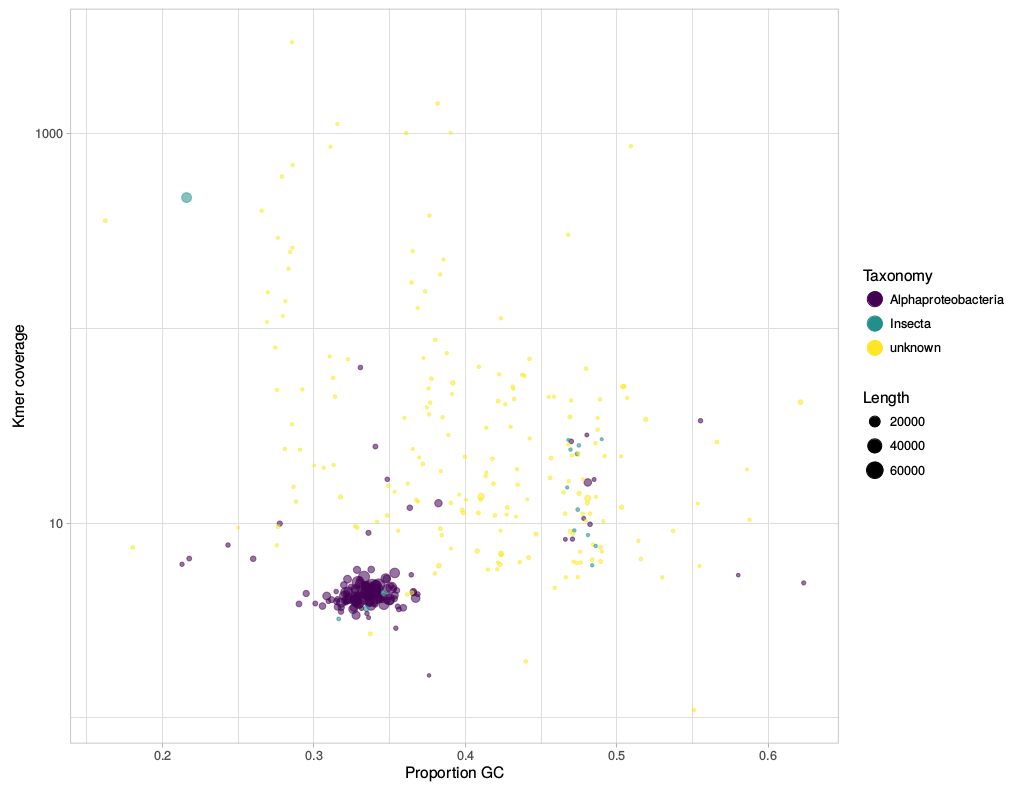 I have written a small script that automates the download of fastq files from the European Nucleotide Archive (ENA). This was created because I was annoyed with the speed NCBI's sra-tools. It takes NCBI SRA accession numbers as input and downloads the fastq files directly from the ENA using wget. The download speed is thus basically only limited by your bandwidth. Any feedback is very welcome!
Find it on the resources page or on github. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed