|
I have officially joined the German Centre for Integrative Biodiversity Research (iDiv)
and Martin Luther University of Halle-Wittenberg as Junior Research Group Leader. The Symbiont Evolution group will be funded by DFG's Emmy Noether Programme (see my previous post). Our research will explore symbiont host shifts using Spiroplasma/Drosophila and Wolbachia/solitary bees as model symbiont/host systems. Our work will include experimental evolution as well as sampling of natural symbiont populations, and of course lots of genomics. If any of that sounds exciting, I'd love to hear from you! PhD positions will be advertised soon, but there is also lots of room to explore related avenues of research. There are several funding options for independent projects – just get in touch and we can explore possibilities. iDiv is a wonderful and unique place of work with excellent facilities and infrastructure. Learn more about it here.
2 Comments
I'm very happy to announce that the DFG will fund my project "What makes a symbiont succeed? The role of host shifts in arthropod symbiont evolution." under their Emmy Noether Programme. I'll be establishing my group in Germany soon. Stay tuned for more details, including updates on open positions.
Our work on Spiroplasma evolution in Drosophila is now available as a preprint here. This is the first publication from my postdoctoral work with Greg Hurst at the University of Liverpool. It was also a very nice collaboration with great people from Liverpool, Texas A&M and EPFL. The work was funded by the European Union’s Horizon 2020 Research and Innovation Program under Marie Sklodowska-Curie grant agreement 703379.
Spiroplasma is a very interesting symbiont with lots of peculiar features (I'd recommend these reviews about its biology). After some mostly anecdotal evidence that Spiroplasma symbionts evolve quickly, we here have determined Spiroplasma mutational rates systematically. We find that indeed, Spiroplasma can be considered a hypermutator, especially when compared with Wolbachia (the only other natural inherited symbiont of Drosophila). There are some interesting implications for Spiroplasma evolutionary ecology that arise from this which we discuss in the manuscript. We also show and discuss lots of comparative genomics data. If you want to learn more, please have a look at the video below which is a recording of my presentation about this work from the Symbiosis Seminar Series organised by Nicole Gerardo and Greg Hurst. UPDATE (August 13, 2020):
You can still apply for this position until September 14, 2020. Please follow the updated link and get in touch with any questions. (old post below)
A recent paper in Nature Communications (Abadi et al. 2019) investigated model selection in phylogeny reconstruction. Selecting an appropriate model of nucleotide substitution is considered best practice in phylogenetics, and indeed many studies have show that accurate modelling of substitution processes can substantially improve phylogenetic estimates. The authors quite surprisingly find that this practice may not be necessary after all. From multiple datasets of diverse simulated sequences, they find that the models chosen by commonly used criteria do not perform better than the most complex model. They conclude that cases model selection can be skipped altogether, and all phylogenetic inferences be performed with a complex model.
UPDATE (November 25, 2019): What is discussed here, together with some more in depth analyses, are now available as preprint here. There are other interesting aspects of this paper, but I here want to focus on the claim (and that data that's used to support it) that model selection does not improve phylogenetic inference. Below, I will argue that
I have joined the Department of Biological and Medical Sciences at Oxford Brookes University as Lecturer in Ecology and Conservation. I am looking forward to establishing a group here and to teaching in Ecology, Conservation, and Biodiversity.
I want to acknowledge all the people how have been very supportive since I moved to the UK, especially Greg Hurst, who was a great mentor and a lot of fun to work with, and Kayla King, who helped me with the transition to Oxford. Thank you! New research with my input was also published:
This blog post deals with the various ways of downloading large amounts of sequencing data (e.g., from NCBI’s SRA database). When I needed to bulk download short read for a recent project, it took me some time to figure out how to achieve this efficiently, and I am sharing my experience here in the hope it might be useful.
The problem: you want to download lots of sequencing data (typically in form of Illumina generated reads), e.g., to reproduce a published experiment. The amount of data makes it impossible to click+download through a browser interface. There are two potential solutions: 1) download via NCBI’s SRA toolkit, and 2) access ftp servers directly. Before writing about science again (new post is in the making), I have uploaded two scripts that I find useful for my work: gc_cov.pl This script is basically a very simplified version of the blobplots function from the BlobTools package. It creates GC-coverage plots directly from SPAdes assembly files, without the need for mapping the reads back to the assembly. Since I use mainly SPAdes anyway, this has been quite handy. The script will also annotate the plot when a taxonomy file is provided, which can be generated, e.g., from blast outputs. Below is an example for the plots that can be generated with the script. 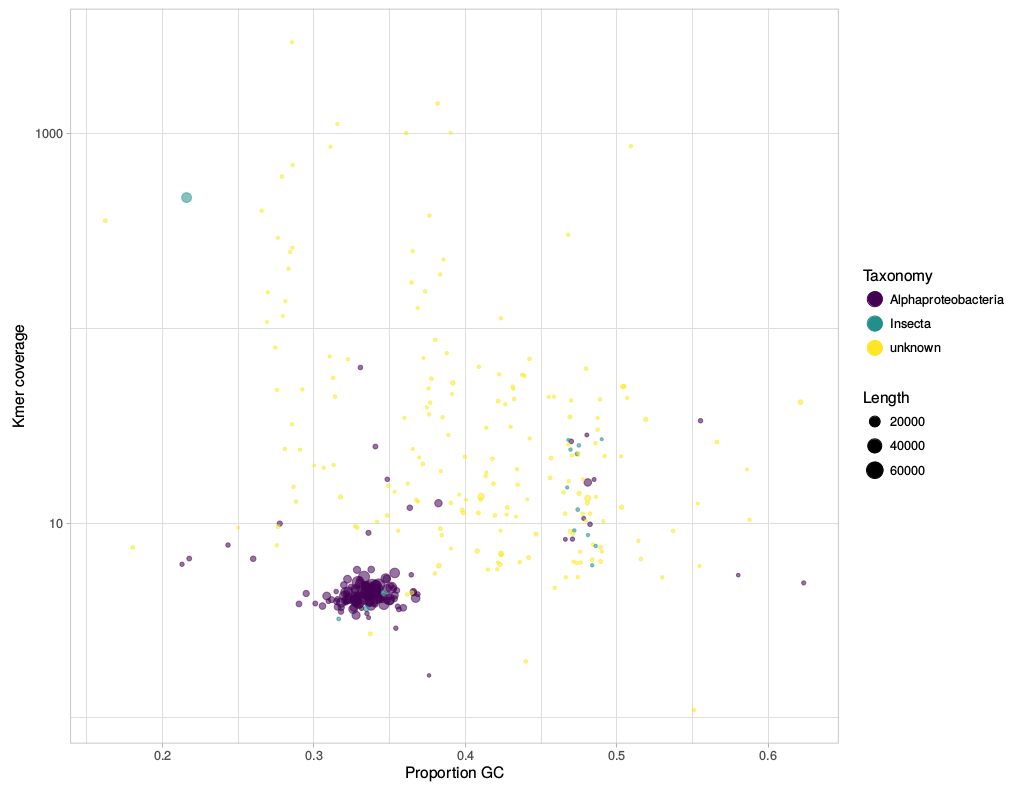 I have written a small script that automates the download of fastq files from the European Nucleotide Archive (ENA). This was created because I was annoyed with the speed NCBI's sra-tools. It takes NCBI SRA accession numbers as input and downloads the fastq files directly from the ENA using wget. The download speed is thus basically only limited by your bandwidth. Any feedback is very welcome!
Find it on the resources page or on github. This post is a ‘behind the paper’ story of our publication ‘Short reads from honey bee (Apis sp.) sequencing projects reflect microbial associate diversity’ which was just published in PeerJ. I will explain the motivation behind the study and also show some new data generated with our approach.
UPDATE (July 24, 2017): Our paper was covered in the "The Molecular Ecologist" blog: http://www.molecularecologist.com/2017/07/genomes-are-coming-sequence-libraries-from-the-honey-bee-reflect-associated-microbial-diversity/ Part one: background & motivation I work as a postdoc in Greg Hurst’s group, who has projects on many different bacterial symbionts (https://sites.google.com/site/hurstlab/home). The project of his PhD student Georgia Drew aims to identify the potential impacts of Arsenophonus on honey bee health (https://eegid.wordpress.com/phd-students/georgia-drew/). This bacterium is an inherited symbiont of arthropods, and has been found in honey bees and other bees occasionally (Aizenberg-Gershtein et al. 2013; Gerth et al. 2015; Yañez et al. 2016; McFrederick et al. 2017). Its exact role in honey bees is unclear, and this is what Georgia studies. I became involved when Greg suggested to extract genomic data of the honey bee associated Arsenophonus from Apis Illumina data stored public databases. In many cases, symbionts are sequenced inadvertently alongside with their hosts, and a number of symbiont genomes have been extracted from sequencing data of their hosts before (e.g., Salzberg et al. 2005; Siozios et al. 2013). There’s plenty of honey bee sequencing data around, so it was definitely worth checking if we could get an Arsenophonus genome ‘for free’, without actually sequencing it ourselves. |
Welcome!This is the website of Michael Gerth. I am a biologist with an interest in insects and the microbes within them. Click here to learn more. Archives
June 2022
Categories
All
|
 RSS Feed
RSS Feed